Hybrid Methods for Large Chromophores in Complex Environments
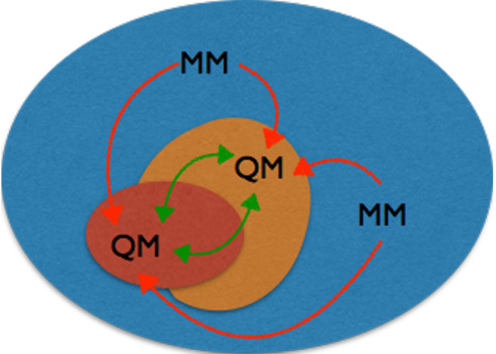
Mutually Polarizable QM/QM Embedding Schemes
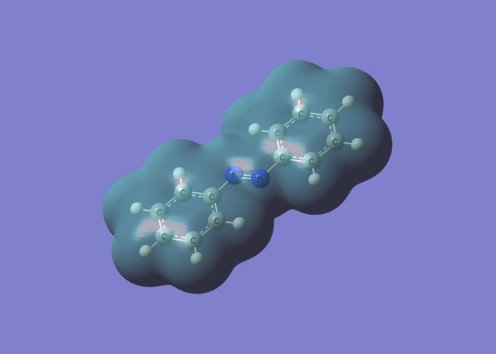
Coupled Cluster Methods and Polarizable Solvation Models
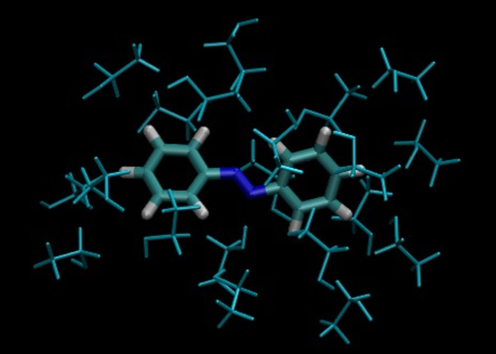
Coupled Cluster Methods and Polarizable Solvation Models, figure #2
Chromphores of interest in real applications are often too large to be treated entirely at a high level of theory. Therefore, hybrid methods that combine a high quantum mechanical (QM) level of theory for the core region, and lower levels for outer layers are necessary to strike the right balance between computational cost and accuracy. We develop hybrid methods that combine accurate QM methods (e.g., from coupled cluster theory) with cheaper QM methods (e.g., from density functional theory) and with polarizable classical models (e.g., implicit solvation models or polarizable force fields) to describe the electronic response properties of large chromophores in complex environments.
When molecules are too large to be treated entirely at an accurate QM level, hybrid methods are often employed to reduce the computational cost. In hybrid methods, the system is separated in layers where the most important region is treated with a high level of theory while the rest is treated at a lower, more manageable level of theory. The success of this approach is often connected with the ability to treat the interaction between the layers appropriately. This becomes even more important when we want to study electronic excited states because of the more polarizable nature of the electron density.
We are working on developing methods to account for mutual polarization between high and low levels of theory in a self-consistent manner, see scheme in Figure. Starting from the ONIOM (Our own N-layer molecular Orbital molecular Mechanics) method of Morokuma for excitation energies, we are working on defining polarizable embedding fields whose parameters are computed on-the-fly from the low-level calculation. Additionally, we are developing techniques to extrapolate multiple excited states simultaneously to avoid the problem of state-matching between different levels of theory.
We also develop strategies to couple CC methods with these solvation models, aiming at the best compromise between accuracy and computational effort. We consider both implicit and explicit polarizable solvation models, shown in Figure, for ground and excited state molecular properties. Our goal is to take advantage of the high-quality of the CC results with the computational efficiency of these solvation models to simulate the spectra of complex chromophores in bulk solution or at the interface between solid and liquid phase.
S. Ren, F. Lipparini, B. Mennucci, M. Caricato*, Coupled Cluster Theory with Induced Dipole Polarizable Embedding for Ground and Excited States; J. Chem. Theory Comput., 15, (2019) 4485.
M. Caricato*, CCSD-PCM Excited State Energy Gradients with the Linear Response Singles Approximation to Study the Photochemistry of Molecules in Solution; ChemPhotoChem, 3, (2019) 1.
M. Caricato*, Linear response coupled cluster theory with the polarizable continuum model within the singles approximation for the solvent response; J. Chem. Phys., 148, (2018) 134113.
S. Ren, J. Harms, M. Caricato*, An EOM-CCSD-PCM Benchmark for Electronic Excitation Energies of Solvated Molecules; J. Chem. Theory Comput., 13, (2017) 117.
A. Biancardi, J. Barnes, M. Caricato*, Point charge embedding for ONIOM excited states calculations; J. Chem. Phys., 145, (2016) 224109.
S. Ren, M. Caricato*, Multi-state extrapolation of UV/Vis absorption spectra with QM/QM hybrid methods; J. Chem. Phys., 144, (2016) 184102.